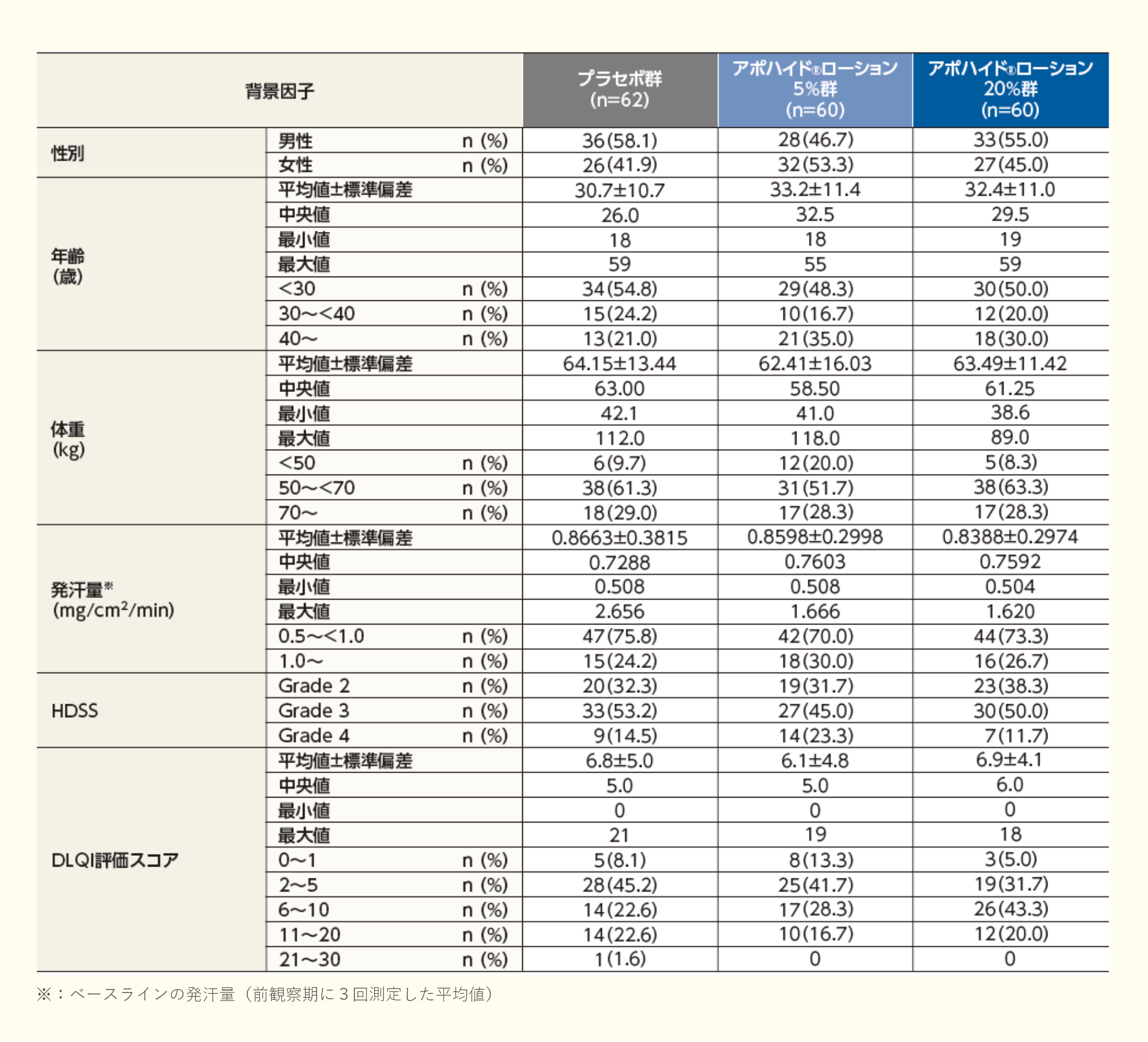
アポハイド®ローション20%(以下、本剤)は、オキシブチニン塩酸塩を有効成分とする久光製薬株式会社(以下、当社)が開発した原発性手掌多汗症治療剤である。
原発性局所多汗症は、頭部・顔面、手掌、足底、腋窩に温熱や精神的負荷の有無いかんに関わらず、日常生活に支障をきたすほどの大量の発汗を生じる状態と定義され、手掌部に発現する原発性局所多汗症を原発性手掌多汗症という1)。本邦における原発性手掌多汗症の有病率は5.33%、平均発症年齢は13.8歳(男性15.0歳、女性11.6歳)であり2)、幼少児期又は思春期頃に発症することが多く1)、その罹患により、学習効率や労働生産性の低下、精神的苦痛、対人関係への悪影響に苛まれ、患者の生活の質(Quality of Life:QOL)の低下をきたすとされている3)。
「原発性局所多汗症診療ガイドライン 2015年改訂版」において、原発性手掌多汗症に対する第一選択の治療法は、塩化アルミニウム外用療法及びイオントフォレーシスとされている1)。しかし、塩化アルミニウムは保険診療に適用のある外用剤がなく院内製剤として処方されていること、イオントフォレーシスは患者自身による機器の購入又は専用の機器を有する医療機関への通院が必要なこと等から、本疾患において、保険診療に適用のある利便性の高い新たな外用剤が望まれていた。
汗腺の一つであるエクリン汗腺は全身に分布しており、エクリン汗腺に存在するムスカリンM3受容体が刺激されると発汗が惹起される4)。本剤の有効成分であるオキシブチニン塩酸塩は、1963年に合成された化合物で、ムスカリンM3受容体にオキシブチニンが結合することで(in vitro)5)、抗コリン作用を有することが確認されている(in vitro)6-10)。また、オキシブチニン塩酸塩の臨床効果には、その活性代謝物であるN-デスエチルオキシブチニン(N-Desethyloxybutynin:DEO)も関与することが示唆されており(in vitro)8,9)、これらによる発汗抑制作用が期待されたため、当社は原発性手掌多汗症を適応とした本剤の開発に着手した。
本剤の臨床開発は、原発性手掌多汗症患者を対象とした国内第Ⅱ相試験(プラセボ対照二重盲検比較試験:03試験)、国内第Ⅲ相試験(プラセボ対照二重盲検比較試験:04試験)、国内第Ⅲ相長期投与試験(長期投与試験:05試験)において有効性及び安全性が確認され、本剤は2023年3月に「原発性手掌多汗症」の効能又は効果で製造販売承認を取得した。
1) 藤本智子 ほか: 日皮会誌 2015; 125(7): 1379-400.
2) Fujimoto T, et al.: J Dermatol 2013; 40(11): 886-90.
3) Hamm H, et al.: Dermatology 2006; 212(4): 343-53.
4) 岩瀬敏 ほか: 日皮会誌 2014; 124(7): 1277-82.
5) Maruyama S, et al.: J Urol 2006; 175(1): 365-9.
6) Noronha-Blob L, et al.: J Pharmacol Exp Ther 1991; 256(2): 562-7.
7) Uchida M, et al.: J Pharmacol Sci 2004; 94(2): 122-8.
8) Mizushima H, et al.: Biol Pharm Bull 2007; 30(5): 955-62.
9) Smith ER, et al.: Arzneimittelforschung 1998; 48(10): 1012-8.
10) Waldeck K, et al.: J Urol 1997; 157(3): 1093-7.

4.5mL(4.32g)(プラスチック容器)×10本
4.5mL(4.32g)(プラスチック容器)×20本
18mL(17.28g)(プラスチック容器)×4本
久光製薬社内資料. アポハイド®ローション承認時評価資料, 原発性手掌多汗症患者を対象とした第Ⅱ相試験.
FAS:Full Analysis Set MMRM:Mixed Modeling for Repeated Measures SOC:System Organ Class PT:Preferred Term

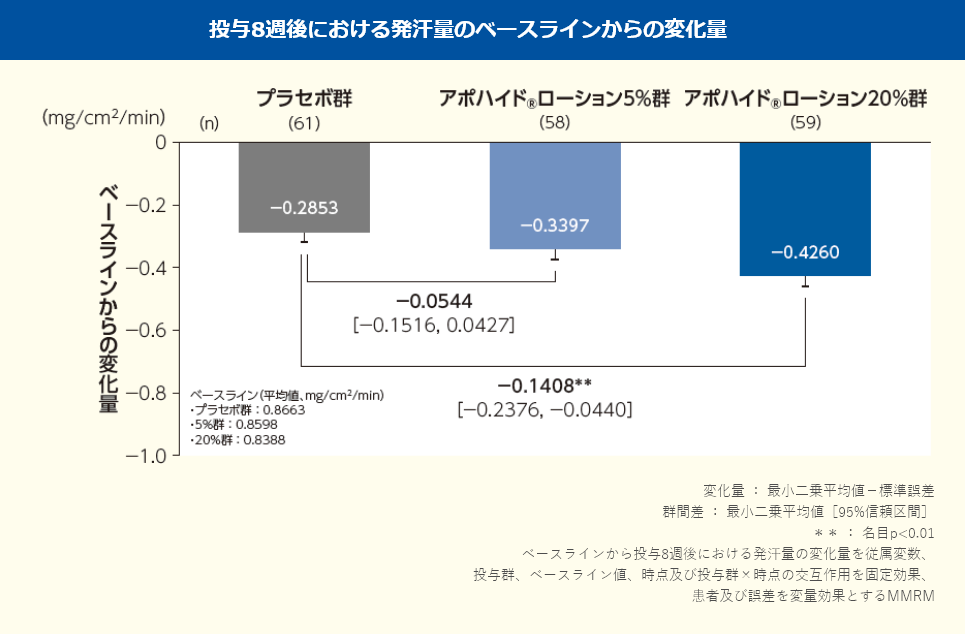
投与8週後における発汗量のベースラインからの変化量(最小二乗平均値)は、プラセボ群で-0.2853mg/cm2/min、アポハイド®ローション5%群で-0.3397mg/cm2/min、20%群で-0.4260mg/cm2/minであり、プラセボ群と比較してアポハイド®ローション5%群では改善が認められなかったが(群間差:-0.0544mg/cm2 /min;名目p=0.2704、MMRM)、20%群では改善が認められた(群間差:-0.1408mg/cm2/min;名目p=0.0046、MMRM)。
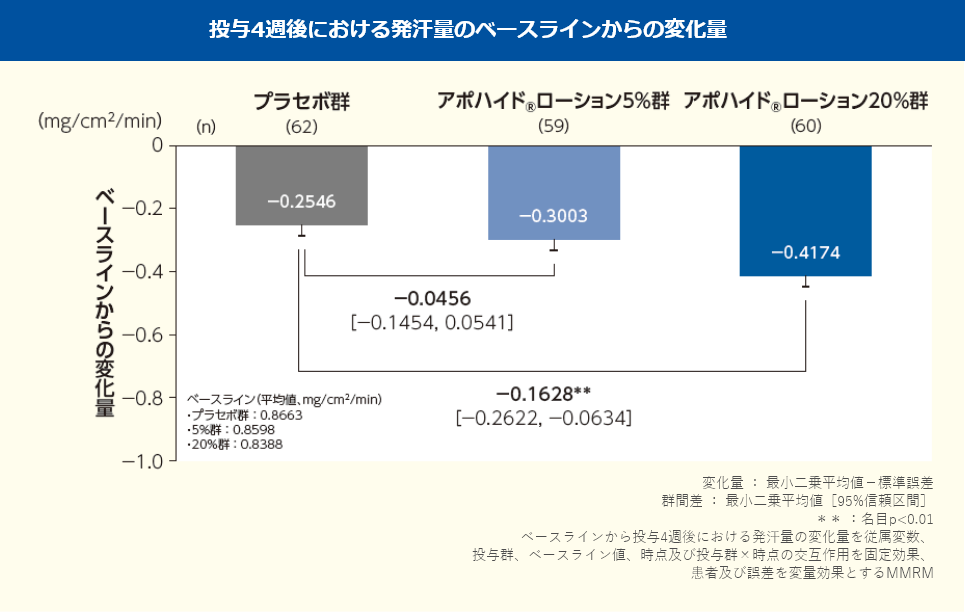
投与4週後における発汗量のベースラインからの変化量(最小二乗平均値)は、プラセボ群で-0.2546mg/cm2/min、アポハイド®ローション5%群で-0.3003mg/cm2/min、20%群で-0.4174mg/cm2/minであり、プラセボ群と比較してアポハイド®ローション5%群では改善が認められなかったが(群間差:-0.0456mg/cm2 /min;名目p=0.3678、MMRM)、20%群では改善が認められた(群間差:-0.1628mg/cm2/min;名目p=0.0015、MMRM)。
久光製薬社内資料. アポハイド®ローション承認時評価資料, 原発性手掌多汗症患者を対象とした第Ⅲ相試験.
FAS:Full Analysis Set SOC:System Organ Class PT:Preferred Term

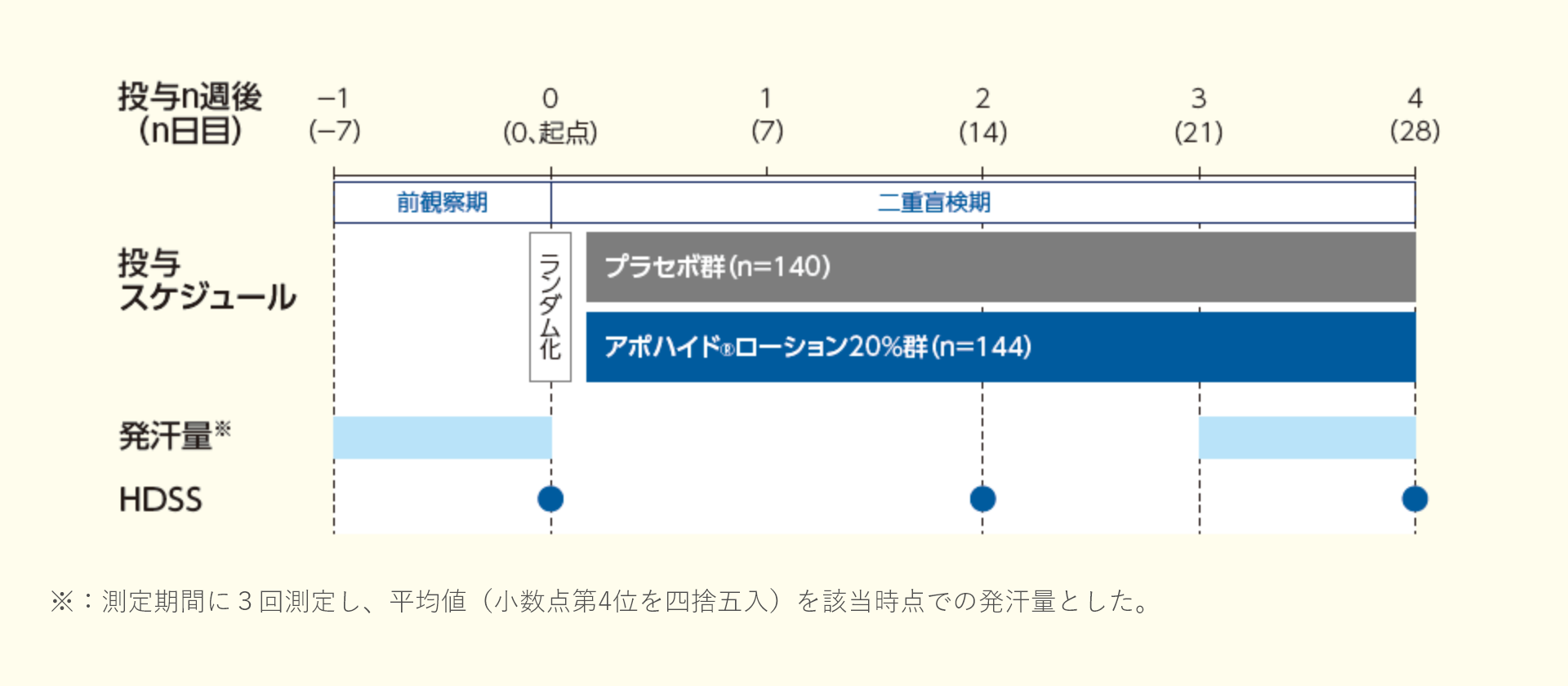
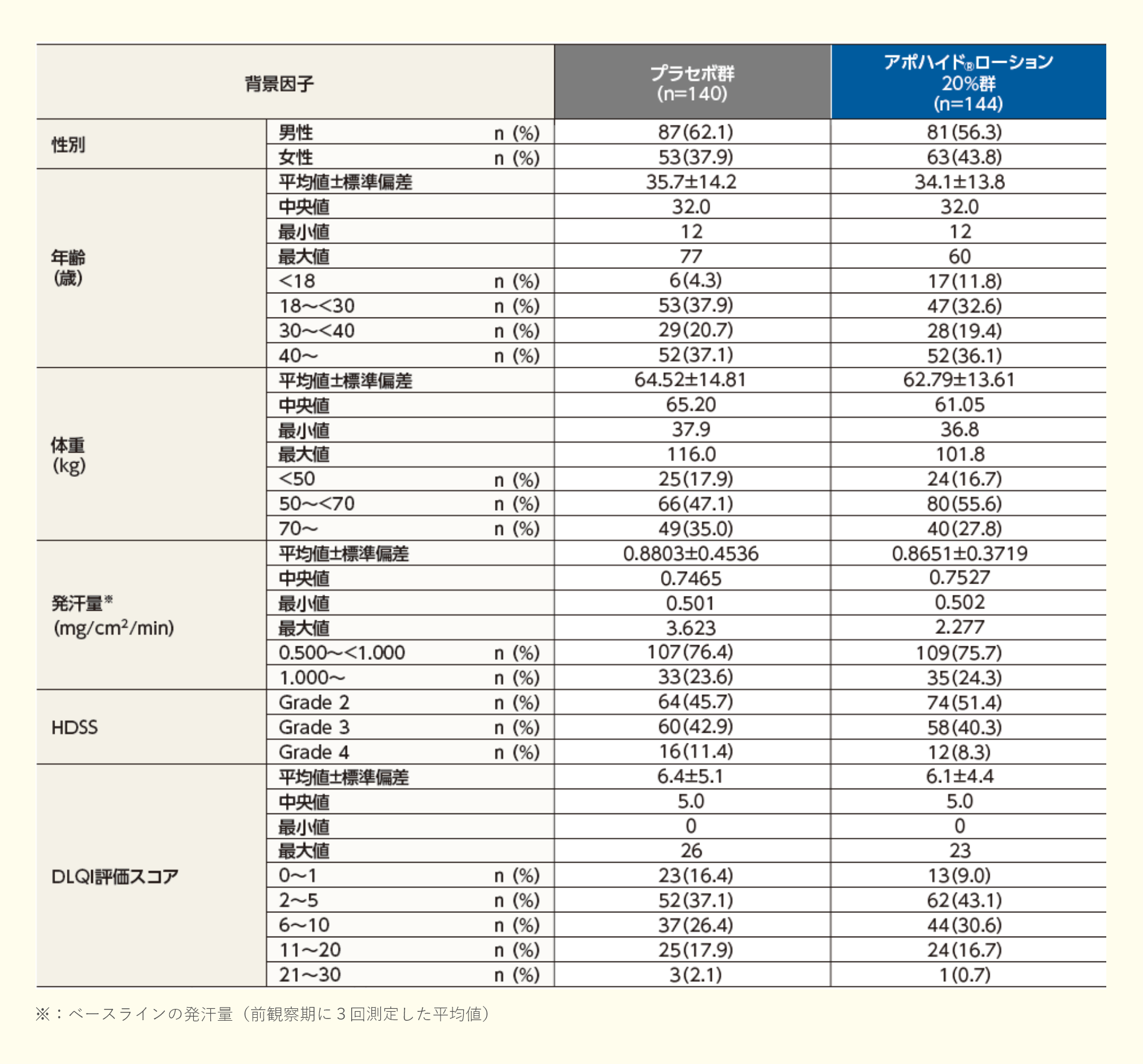
投与4週後における発汗量のレスポンダーの割合は、プラセボ群で24.3%、アポハイド®ローション20%群で52.8%であり、プラセボ群と比較してアポハイド®ローション20%群で有意に高く、プラセボ群に対する優越性が検証された(群間差:28.5%;p<0.001、Fisherの直接確率法)。
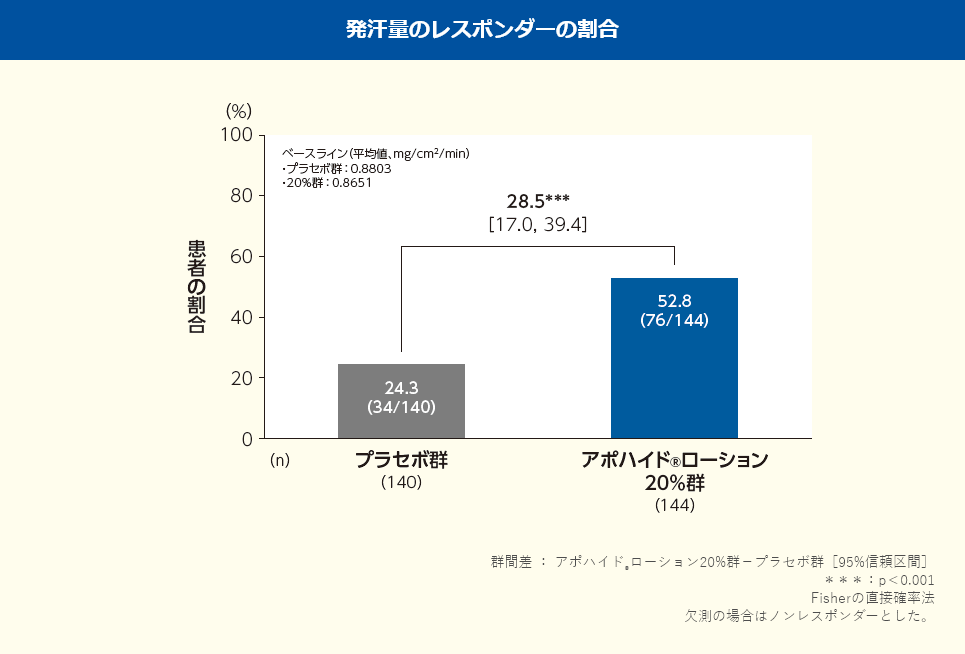
投与4週後における発汗量のベースラインからの変化率(最小二乗平均値)は、プラセボ群で-26.60%、アポハイド®ローション20%群で-48.57%であり、プラセボ群と比較してアポハイド®ローション20%群で改善が認められた(群間差:-21.97%;名目p<0.001、共分散分析)。
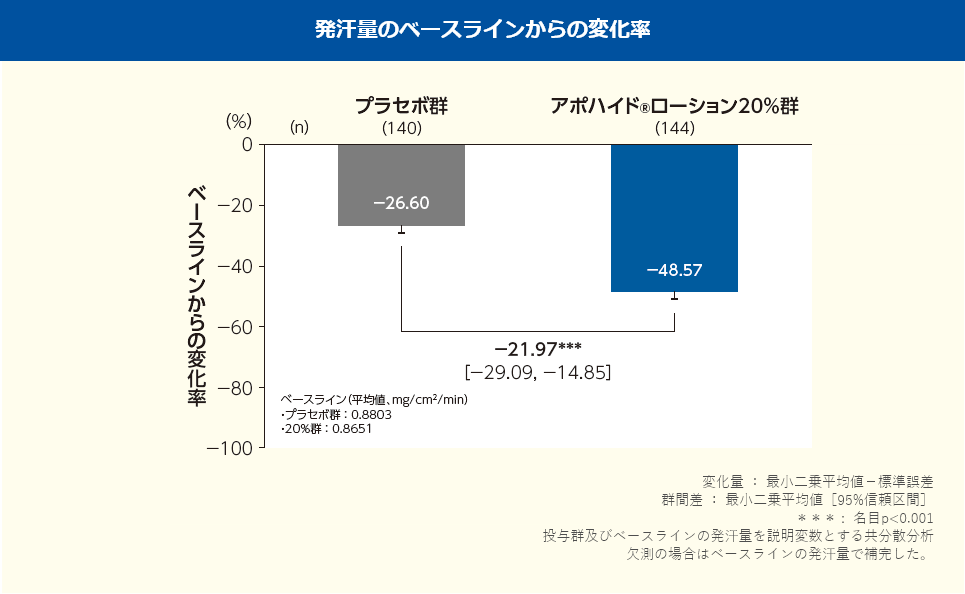
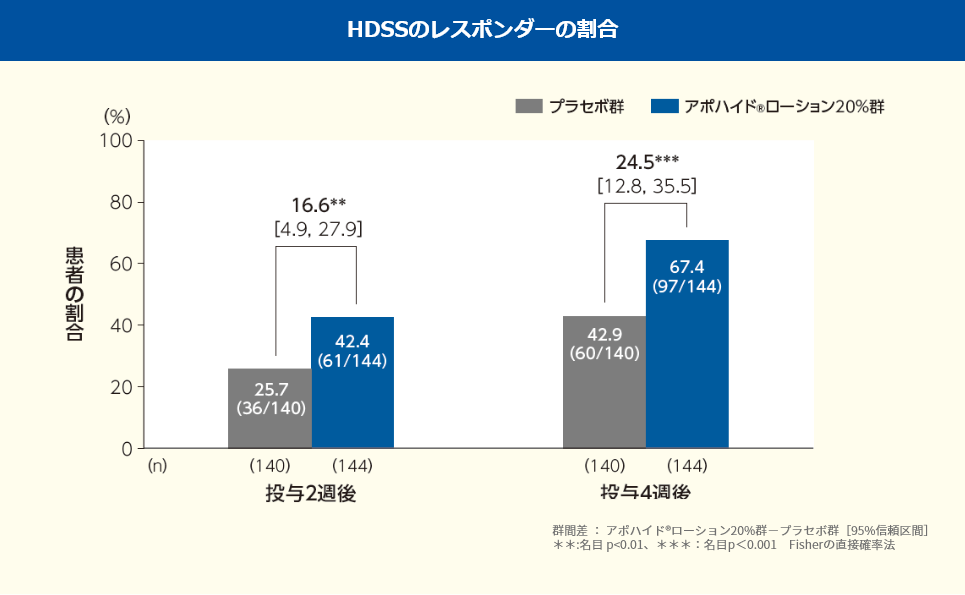
投与2、4週後におけるHDSSのレスポンダーの割合は、プラセボ群ではそれぞれ25.7%、42.9%、アポハイド®ローション20%群ではそれぞれ42.4%、67.4%であり、プラセボ群と比較してアポハイド®ローション20%群ではいずれの時点においても改善が認められた(群間差:投与2週後16.6%、4週後24.5%;名目p=0.0039、名目p<0.001、Fisherの直接確率法)。
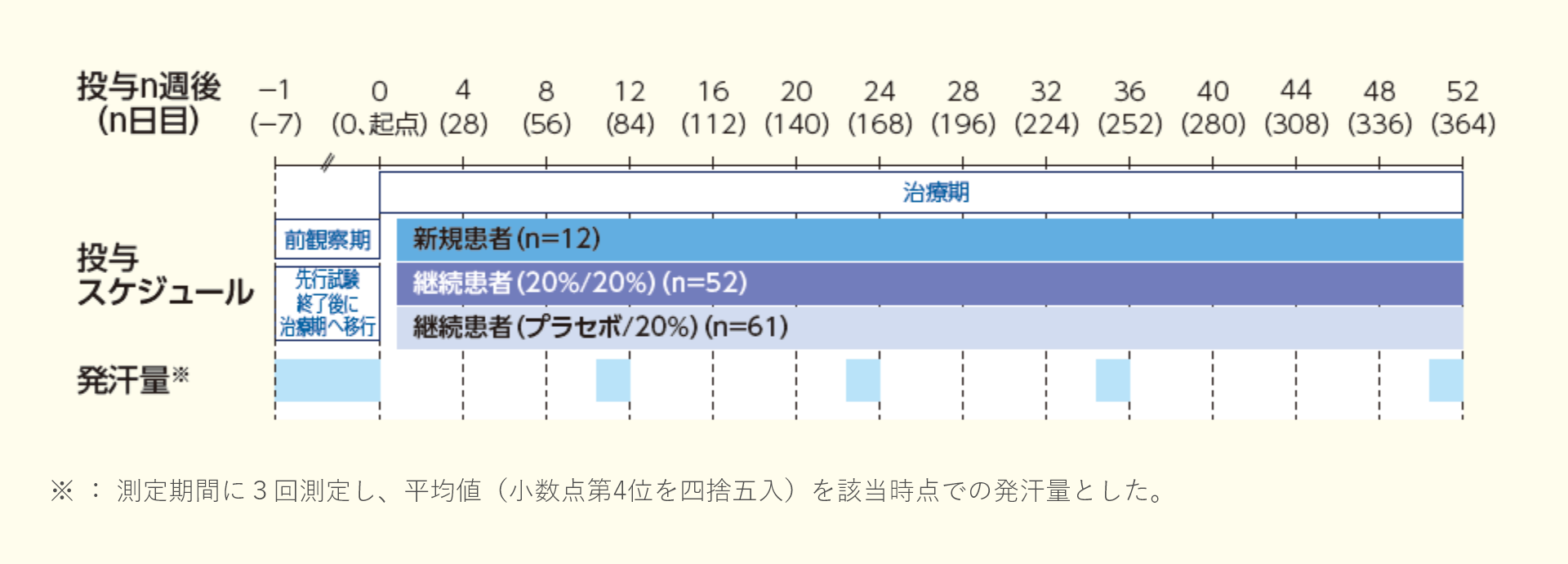
久光製薬社内資料. アポハイド®ローション承認時評価資料, 原発性手掌多汗症患者を対象とした長期投与試験.
※1:国内第Ⅲ相試験(原発性手掌多汗症患者を対象としたプラセボ対照二重盲検比較試験 : 04試験)を完了し、本試験に継続して参加した患者(本試験の治療期から参加)
※2:継続患者は先行試験で基準を満たしているため、新規患者のみの基準
FAS:Full Analysis Set SOC:System Organ Class PT:Preferred Term
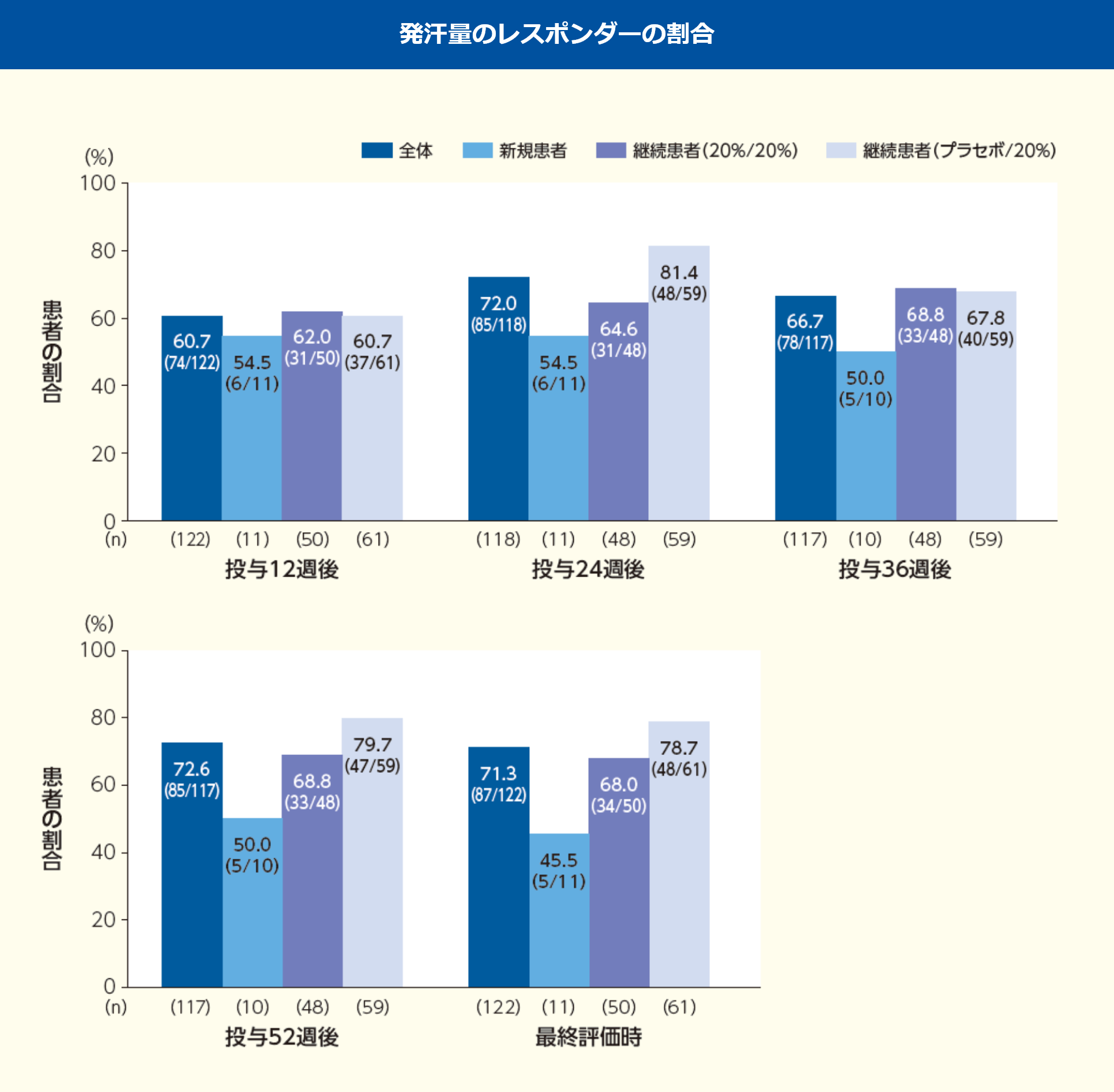
※:投与12, 24, 36, 52週後及び最終評価時
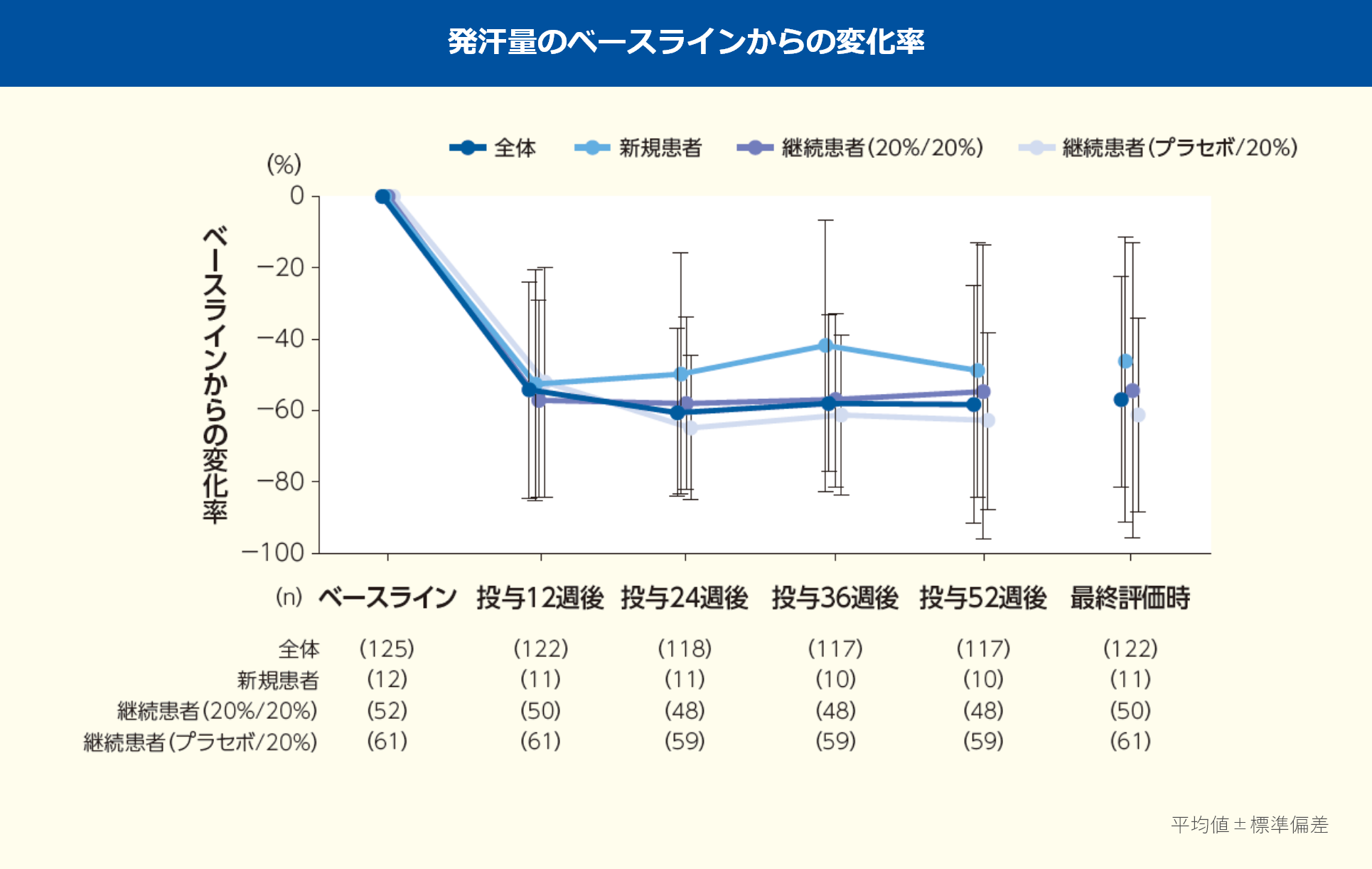
発汗量のレスポンダーの割合(全体)は、投与12週後60.7%、24週後72.0%、36週後66.7%、52週後72.6%、最終評価時71.3%であった。
※:投与12, 24, 36, 52週後及び最終評価時
発汗量のベースラインからの変化率(平均値、全体)は、投与12週後-54.05%、24週後-60.47%、36週後-57.84%、52週後-58.22%、最終評価時-56.83%であった。
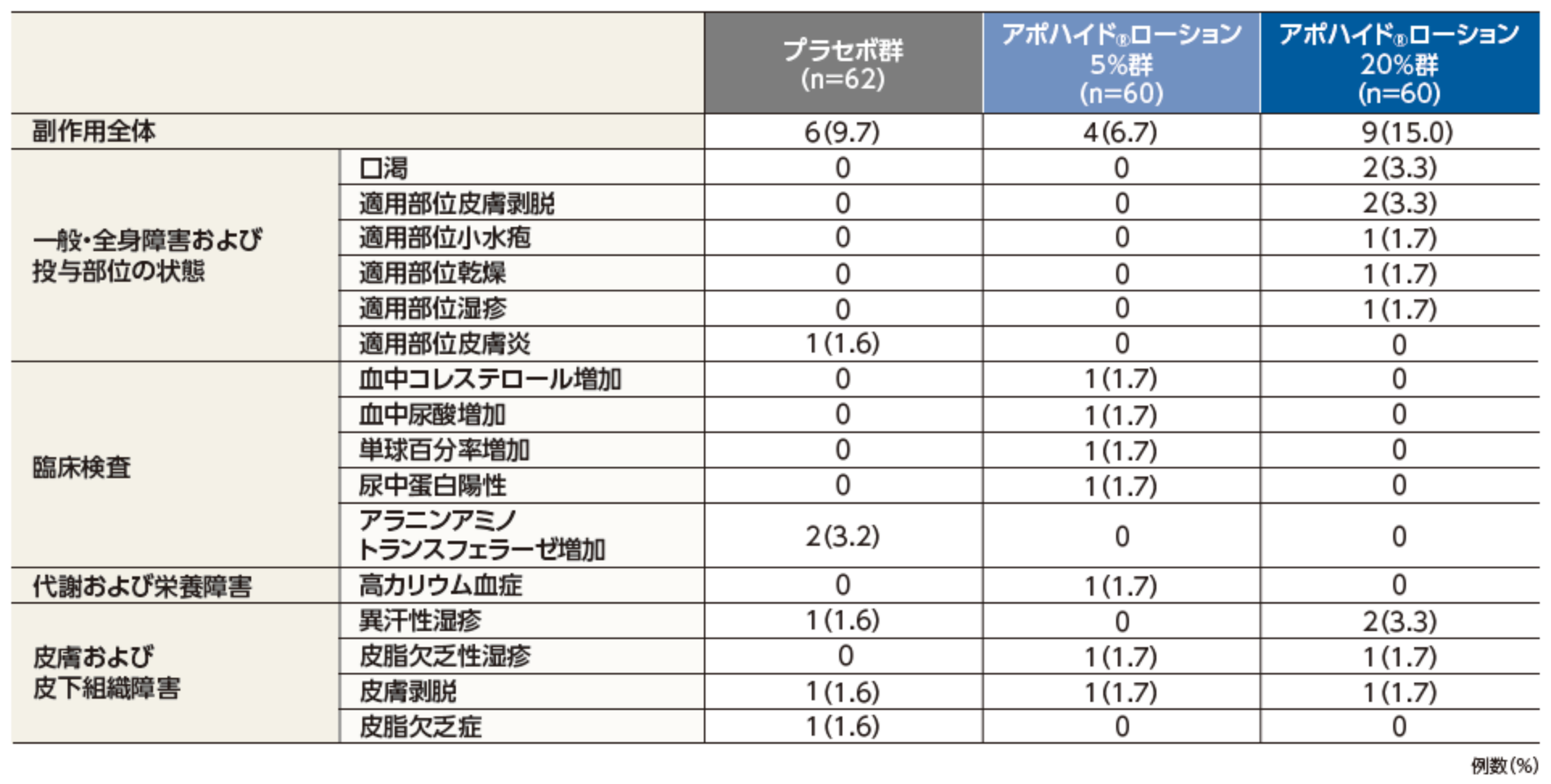
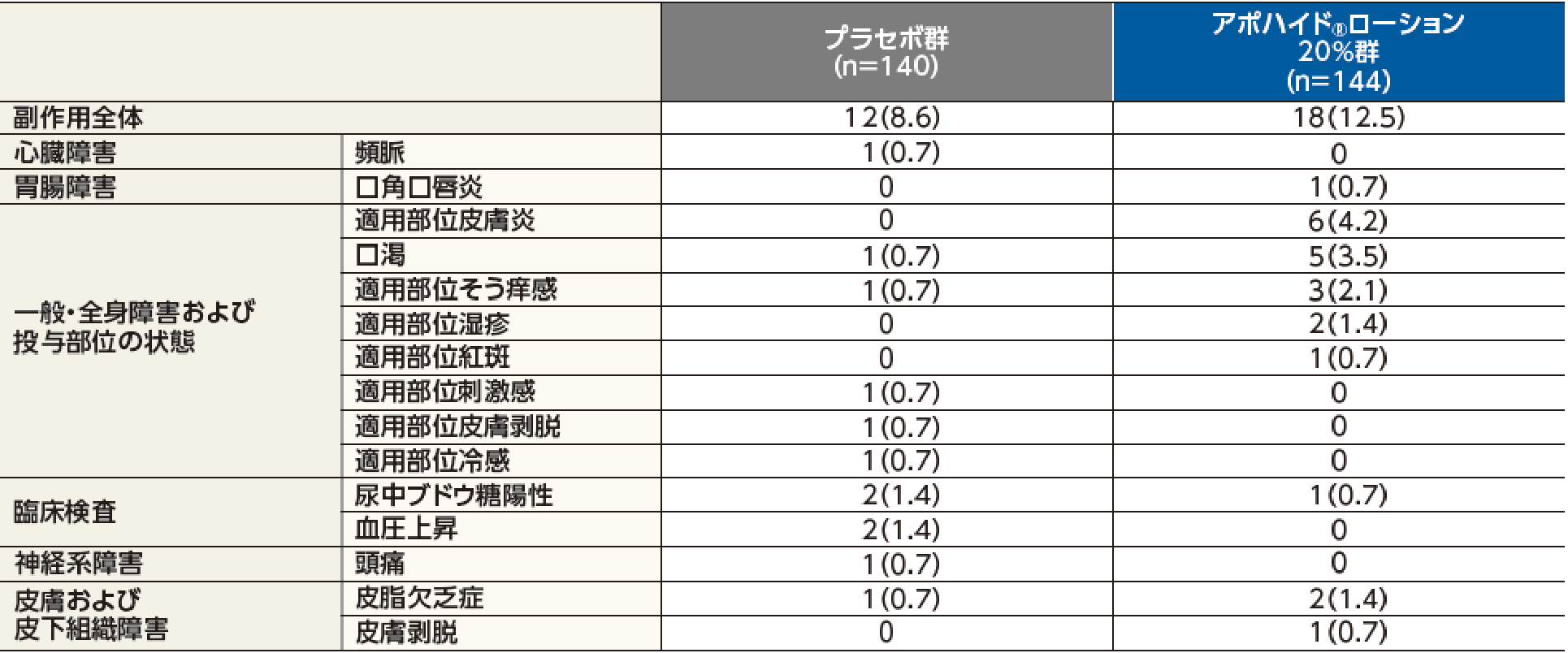
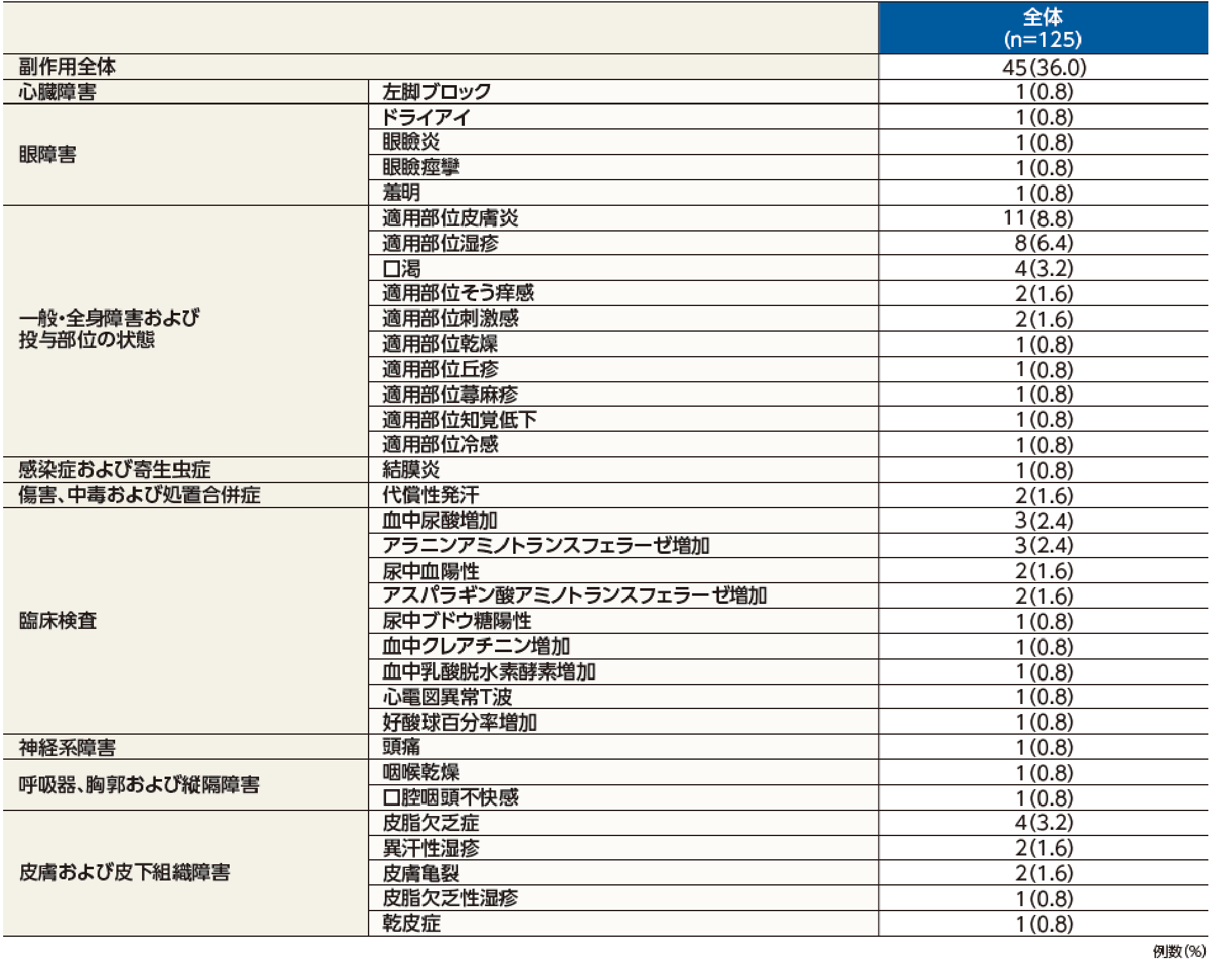
副作用発現率は36.0%(45/125例)で、主な副作用は、適用部位皮膚炎8.8%(11/125例)、適用部位湿疹6.4%(8/125例)、口渇、皮脂欠乏症が各3.2%(4/125例)であった。
投与中止に至った副作用は、適用部位皮膚炎が2例であった。
本試験において、重篤な副作用及び死亡例は認められなかった。
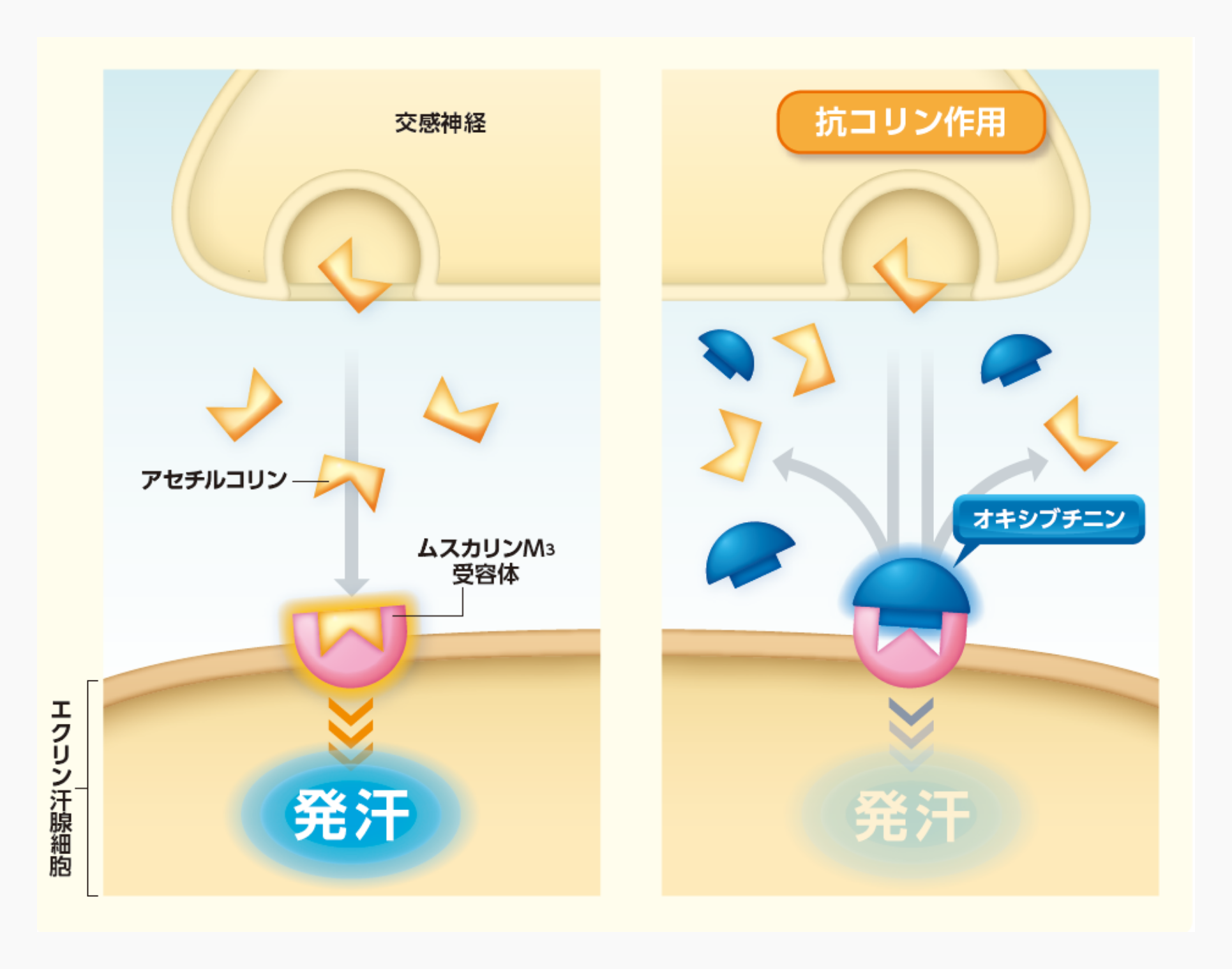
オキシブチニン塩酸塩は、エクリン汗腺に発現するムスカリン受容体にオキシブチニンが結合することで抗コリン作用を有することにより、抑汗作用を示すと考えられている。
汗腺の一つであるエクリン汗腺は全身に分布しており、エクリン汗腺に存在するムスカリンM3受容体が刺激されると発汗が惹起される1)。オキシブチニンはムスカリンM3受容体に対して親和性を有し(in vitro)2)、抗コリン作用を有することが確認された(in vitro)3-7)。
1) 岩瀬敏 ほか: 日皮会誌 2014; 124(7): 1277-82.
2) Maruyama S, et al.: J Urol 2006; 175(1): 365-9.
3) Noronha-Blob L, et al.: J Pharmacol Exp Ther 1991; 256(2): 562-7.
4) Uchida M, et al.: J Pharmacol Sci 2004; 94(2): 122-8.
5) Mizushima H, et al.: Biol Pharm Bull 2007; 30(5): 955-62.
6) Smith ER, et al.: Arzneimittelforschung 1998; 48(10): 1012-8.
7) Waldeck K, et al.: J Urol 1997; 157(3): 1093-7.
より作図
監修 : 埼玉医科大学病院 脳神経内科・脳卒中内科 教授 中里 良彦先生
ヒトムスカリン受容体(M1、M2、M3、M4、M5)を用いた結合実験において、オキシブチニンは[3H]N-メチルスコポラミンの結合を競合的に阻害し、ムスカリンM3及びM4受容体に対して高い親和性を示す傾向がみられた。

平均値±標準誤差
【試験方法】
ヒトムスカリン受容体(M1、M2、M3、M4及びM5)を発現させたチャイニーズハムスター卵巣由来細胞(CHO-K1)細胞膜画分を用いて、[3H]N-メチルスコポラミンの結合に対するオキシブチニン及びDEOの50%阻害濃度(IC50)からKi値※を算出し、ムスカリン受容体に対する親和性を検討した。
※Ki値:ムスカリン受容体に対する親和性([3H]N-メチルスコポラミンに対する競合阻害作用)を示す阻害定数であり、値が小さいほど親和性が高いことを表す。
Maruyama S, et al.: J Urol 2006; 175(1): 365-9.(一部抜粋)
ウサギ、モルモット、ラット及びヒトの摘出組織を用いた実験において、オキシブチニンは抗コリン作用を示した。
※pA2値:用量反応曲線を2倍右方に平行移動させる濃度の負対数。
1) Noronha-Blob L, et al.: J Pharmacol Exp Ther 1991; 256(2): 562-7.
2) Uchida M, et al.: J Pharmacol Sci 2004; 94(2): 122-8.
3) Mizushima H, et al.: Biol Pharm Bull 2007; 30(5): 955-62.
4) Smith ER, et al.: Arzneimittelforschung 1998; 48(10): 1012-8.
5) Waldeck K, et al.: J Urol 1997; 157(3): 1093-7.
健康成人男性にアポハイド®ローション20% 500μLを1日1回8時間、両手掌部に14日間反復経皮投与したときのオキシブチニン及びDEOの血漿中濃度及び薬物動態パラメータは以下の推移を示した。
反復投与時において、血漿中オキシブチニン濃度は投与後72時間(投与3回目)までに、血漿中DEO濃度は投与後168時間(投与7回目)までに定常状態に達すると考えられた。
なお、アポハイド®ローション20%塗布時の全身曝露量は、オキシブチニン塩酸塩経口剤3mg単回投与時の全身曝露量を超えることがある。
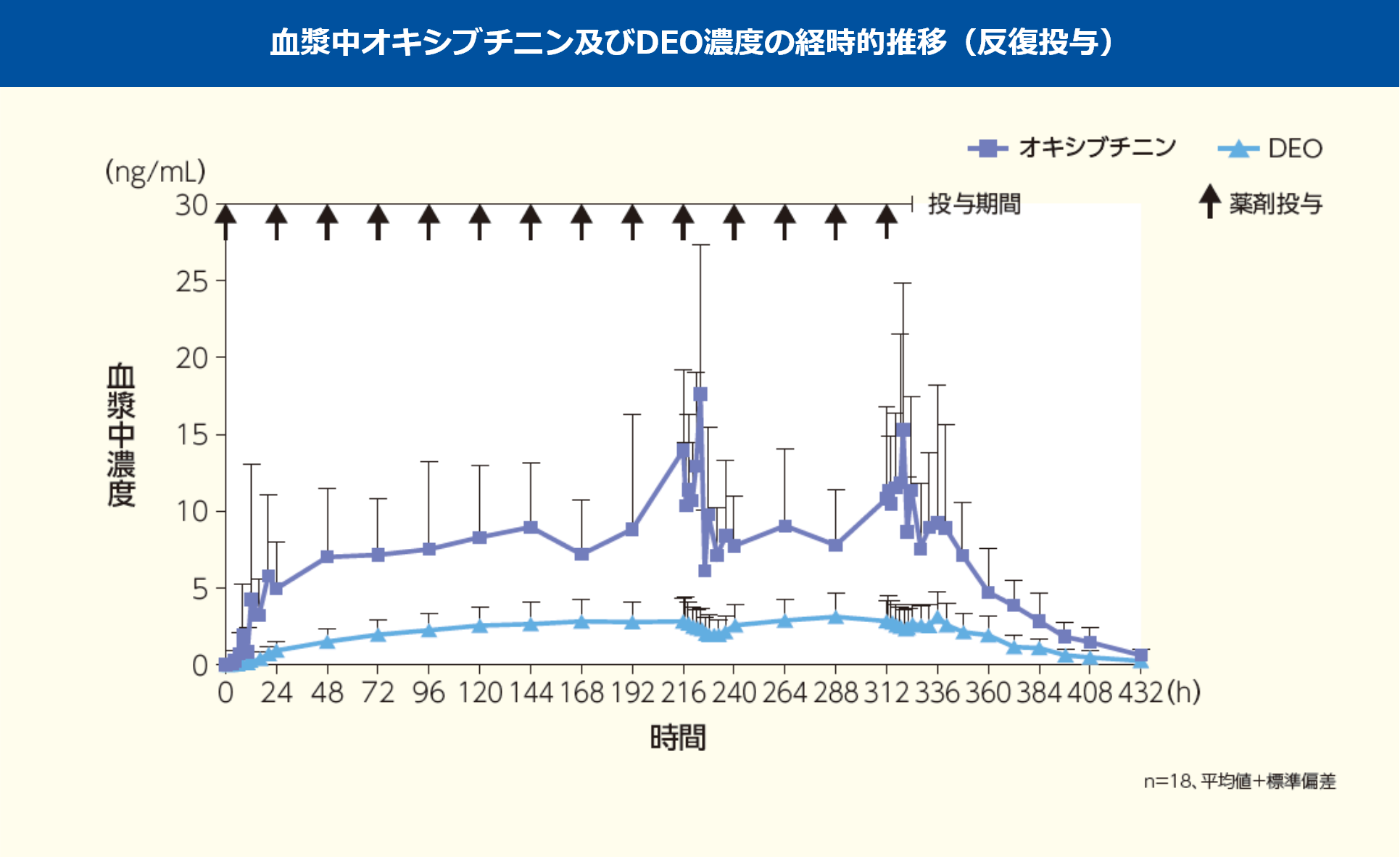
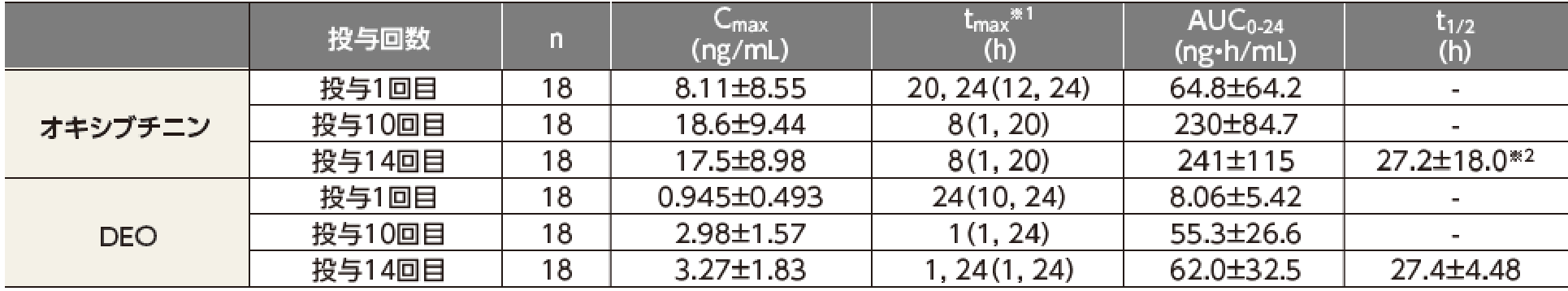
平均値±標準誤差
※1:最頻値(最小値, 最大値)、オキシブチニンの投与1回目、DEOの投与14回目は最頻値が2つ
※2:n=17
【試験方法】
健康成人男性18例を対象に、アポハイド®ローション20%を1日1回、500μL(オキシブチニン塩酸塩として96mgを含有)、両手掌部に14日間反復経皮投与した。投与後8時間に手洗いし薬剤を除去した。
久光製薬社内資料. アポハイド®ローション承認時評価資料, 健康成人を対象とした反復投与試験.
6. 用法及び用量
1日1回、就寝前に適量を両手掌全体に塗布する。
7. 用法及び用量に関連する注意
1回の塗布量は、両手掌に対しポンプ5押し分を目安とすること。
9.特定の背景を有する患者に関する注意(抜粋)
9.1 合併症・既往歴等のある患者
9.1.8 塗布部位に創傷や湿疹・皮膚炎等がみられる患者
創傷や湿疹、皮膚炎等がある部位への使用は避けること。
体内移行量が増加し、抗コリン作用に基づく副作用があらわれやすくなる可能性がある。
国内第Ⅲ相長期投与試験(原発性手掌多汗症患者を対象とした長期投与試験:05試験)において、原発性手掌多汗症患者にアポハイド®ローション20% 500μLを両手掌部に52週間反復経皮投与したとき、各時点におけるオキシブチニン及びDEOの血漿中濃度は以下の推移を示した。
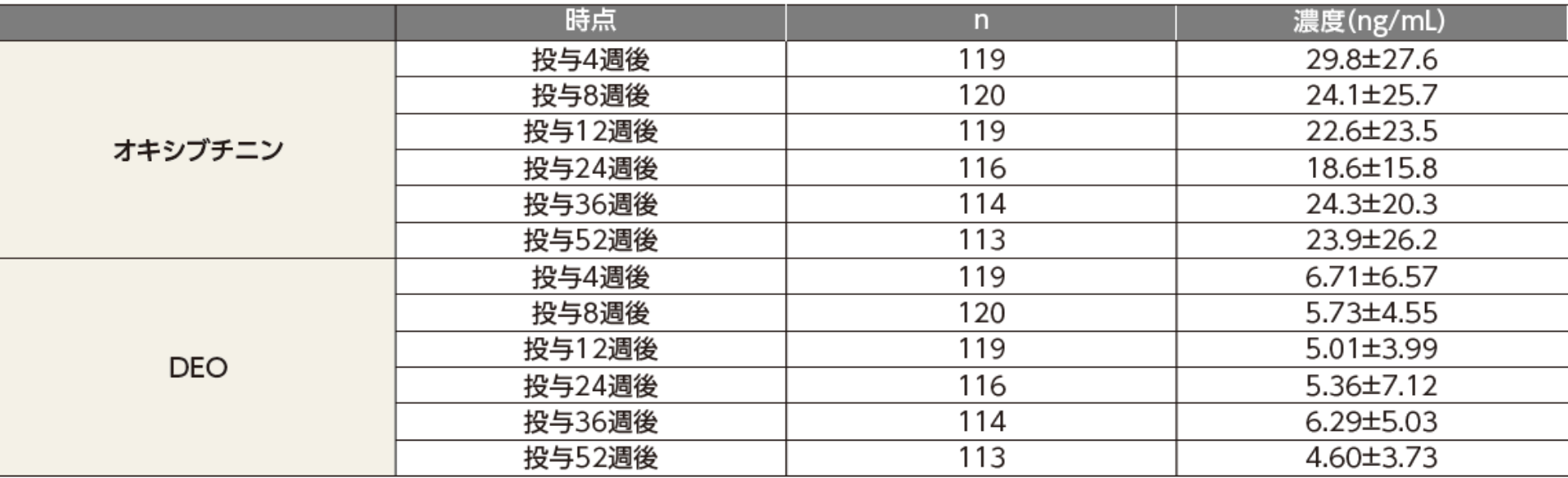
平均値±標準誤差
【試験方法】
原発性手掌多汗症患者125例を対象に、アポハイド®ローション20%を1日1回、就寝前に500μL(オキシブチニン塩酸塩として96mgを含有)、両手掌部に52週間反復経皮投与した。
久光製薬社内資料. アポハイド®ローション承認時評価資料, 原発性手掌多汗症患者を対象とした長期投与試験.
ラットの正常皮膚及び損傷皮膚にアポハイド®ローション20% 10μLを単回経皮投与したとき、血漿中オキシブチニンの濃度推移及び薬物動態パラメータを以下に示した。Cmax及びAUC0-in(f 平均値)は、正常皮膚でそれぞれ4.98ng/mL、229ng・h/mL、損傷皮膚でそれぞれ10.6ng/mL、506ng・h/mLであり、損傷皮膚では正常皮膚のそれぞれ約2.1倍及び約2.2倍を示した。
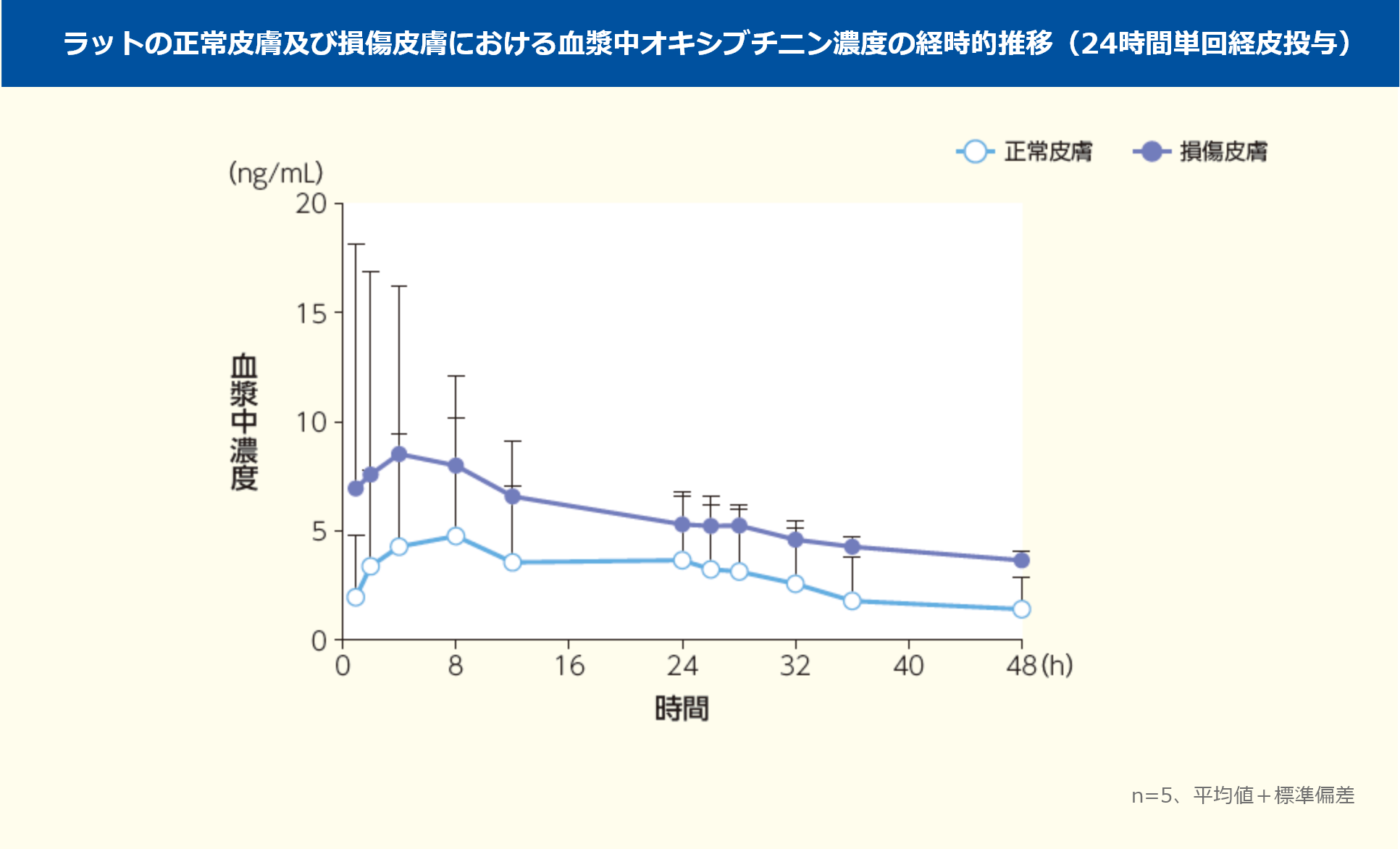

n=5、平均値±標準偏差
【試験方法】
SD系雄性ラット(n=5)の背部正常皮膚及び損傷皮膚に、アポハイド®ローション20%を10μL(オキシブチニン塩酸塩として1.92mgを含有)、24時間単回経皮投与した。
久光製薬社内資料. アポハイド®ローション承認時評価資料, ラットに単回経皮投与後の血漿中濃度.
本剤における薬物吸収後の分布、代謝、排泄、薬物動態学的相互作用については、すでに評価済みである経皮吸収型製剤ネオキシ®テープ73.5mg※と同様と考えられたため、新たな試験は実施しませんでした。本項では同製品の概要を紹介します。
※一般名:オキシブチニン塩酸塩、有効成分:1枚中オキシブチニン塩酸塩73.5mg、承認日:2013年3月25日
<ネオキシ®テープ73.5mgの「6.用法及び用量」>
通常、成人に対し本剤1日1回、1枚(オキシブチニン塩酸塩として73.5mg)を下腹部、腰部又は大腿部のいずれかに貼付し、24時間毎に貼り替える。
ウサギ、モルモット、ラット及びヒトの摘出組織を用いた実験において、オキシブチニンは抗コリン作用を示した。
オキシブチニンは主に肝臓で代謝され、活性代謝物であるDEO等に代謝される。また、ヒト肝ミクロゾームを用いた検討により、オキシブチニンの代謝には主にCYP3A4及びCYP3A5が関与していることが考えられた。
Lukkari E, et al.: Pharmacol Toxicol 1998; 82(4): 161-6.
Yaïch M, et al.: Pharmacogenetics 1998; 8(5): 449-51.
健康成人男性(n=8)にオキシブチニン塩酸塩52.5mgを含有する経皮製剤を下腹部に1日1回7日間反復貼付したとき、貼付開始後144~168時間(貼付7回目)の尿中排泄率(オキシブチニン及び4種の代謝物)は、投与量に対して1.4%であった。また、その内訳は3.8%がフェニルシクロヘキシルグリコール酸、30.8%が4-水酸化DEO、65.4%が4-水酸化フェニルシクロヘキシルグリコール酸であり、オキシブチニン及びDEOはほとんどみられなかった。105mgを含有する経皮製剤貼付時においても同様の傾向が示された。
久光製薬社内資料. 第Ⅰ相反復投与試験(ネオキシ®テープ73.5mg、2013年3月25日承認).
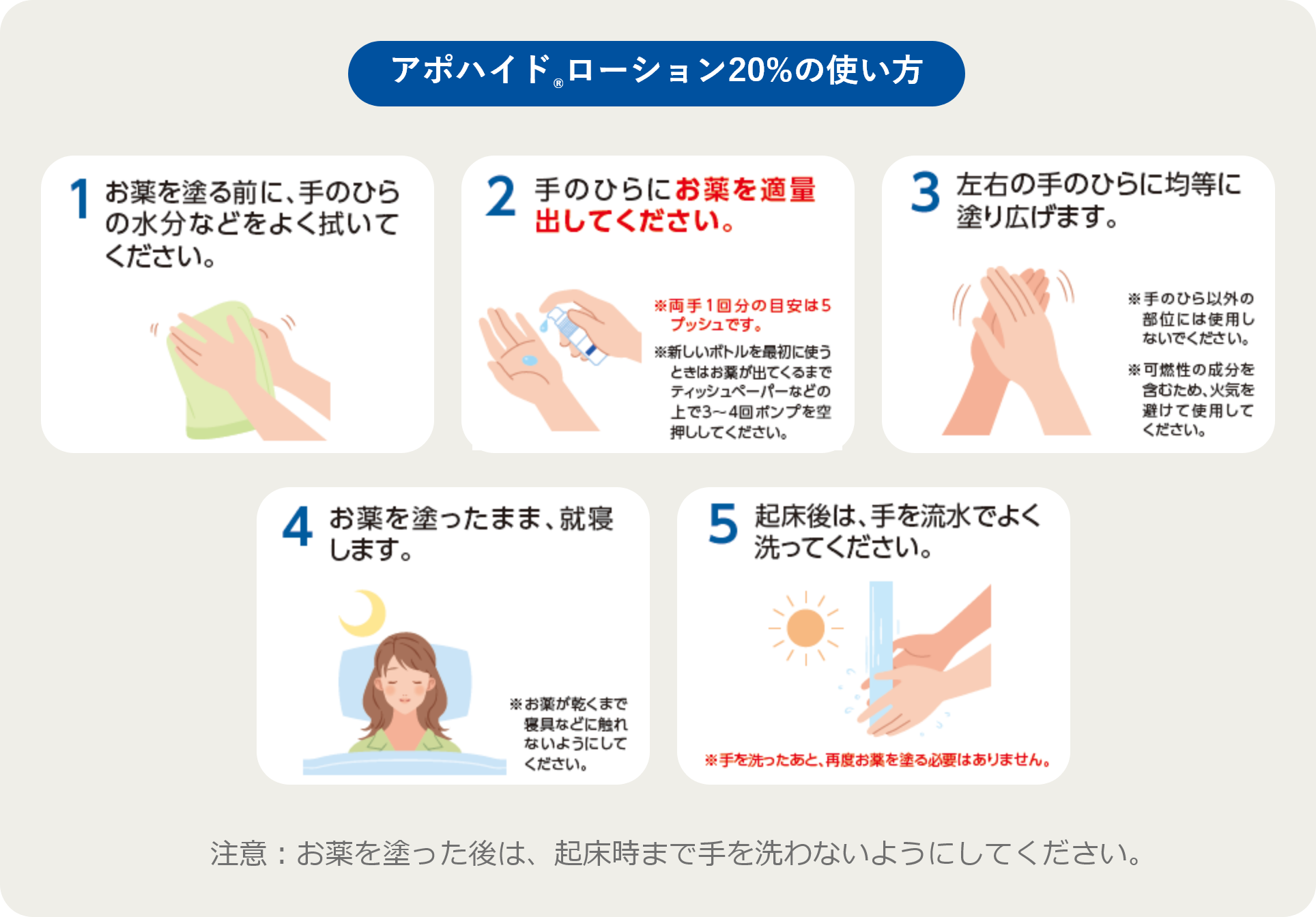
ご使用の際には必ずお読みください。
このお薬は皮膚から吸収されて、手のひらの汗を抑えるお薬です。
1日1回就寝前に使用してください。
子どもの手や目の届かないところに保管してください。

医師または薬剤師の指示に従って使用してください。
子どもの手や目の届かないところに保管してください。
使用するときの注意
ーお薬を塗ったあとの注意ー
起床後に手を洗うまでの間は、以下のようなことに注意してください。
保管上の注意
捨てるときの注意
何か異常が認められた場合には、お薬の使用を中止し、すぐに医師または薬剤師にご相談ください。
ページトップへ戻る皮膚科・形成外科領域の関連コンテンツをご紹介しています。
ぜひ一度ご利用ください。


